Did you know it takes about 5-10 years to train a drug FDA investigator to be proficient? And even with such long and rigorous training, not all FDA investigators are created equal (the human factor). Have you noticed why certain inspections have no FDA-483 while others have significant FDA-483 observations, yes at the same site by different FDA investigators? I had the privilege of being a Level III Pharmaceutical Inspectorate and a Level II Drug Auditor auditing Level 1 FDA drug investigators to become Level II certified FDA investigators in 2010. Even some experienced FDA investigators did not pass FDA drug certification at the time when FDA created this certification program. This shows the importance of continuous training and certifications that the Office of Regulatory Affairs (ORA) has done to ensure that FDA investigators are efficient and thorough when conducting inspections.
At the Center for Drugs Evaluation and Research (CDER) level, not all assessors (previously referred to as reviewers) are created equal as well (the human factor) while assessing companies’ facility and/or chemistry manufacturing and control (CMC) data. This is probably one of the reasons why FDA’s Office of Pharmaceutical Quality (OPQ) announced the FY2025 Experiential Learning Site Visit Program (ELSVP) which invites pharmaceutical companies to participate in site visit proposals with OPQ in order to expose OPQ staff to drug development and manufacturing processes (Federal Register :: Office of Pharmaceutical Quality Experiential Learning Site Visit Program; Program Announcement). Similar to ORA, I believe CDER recognizes the importance of training to providing quality assessment of drug applications and/or manufacturing facilities.
So why is achieving and maintaining quality so challenging? In my view, it largely comes down to human factors, which is why it all starts with training. Making pharmaceuticals is like following a complex recipe —more steps involving human intervention increase the chances of mistakes. Take the visual inspection of sterile injectable products, for example.
Even with rigorous training, medical exams, and visual inspection tests, how can you ensure consistent performance across different operators? The reality is, we’re human, and everyone has good and bad days. It only takes one bad moment to affect quality.
Even with an Acceptance Quality Limit (AQL) in place — where another inspector checks the product — we’re still taking a risk that some patients may receive a product with particulate matter. To reduce this risk, additional checks, like a robotic visual inspection, may be incorporated as a redundancy to catch human errors. However, whilst increasing the use of automated activities and controls can help improving the quality of the output, human beings cannot be replaced in full and they will continue to play an important role in the future for ensuring the manufacturing of safe and effective products.
In addition, control is an inherent and critical step of any successful process. In this sense, inspections, audits and assessments will continue to play a pivotal role in pharmaceutical manufacturing.
What I offer to the industry is the closest opportunity to think like regulators—by training and developing auditors to operate like pseudo-FDA investigators. These auditors will be trained to deliver high-impact FDA-483 like observations, similar to the ones that lead to warning letters, injunctions, consent decrees, or voluntary facility shutdowns when necessary.
This training is paired with an advanced inspection platform powered by artificial intelligence (AI), designed to enhance both the efficiency and quality of audits. I’ve developed global inspection software with AI capabilities, initially focused on the USFDA market but evolving to serve other regulatory regions as well.
The goal of this software is to empower auditors to conduct efficient, accurate, and thorough internal audits, preparing companies for pre-approval and surveillance inspections. This proactive approach ensures readiness for FDA inspections at all times, reducing the need to react to FDA-483 observations after the fact, and at a very high cost for the reputation of the company and its resources.
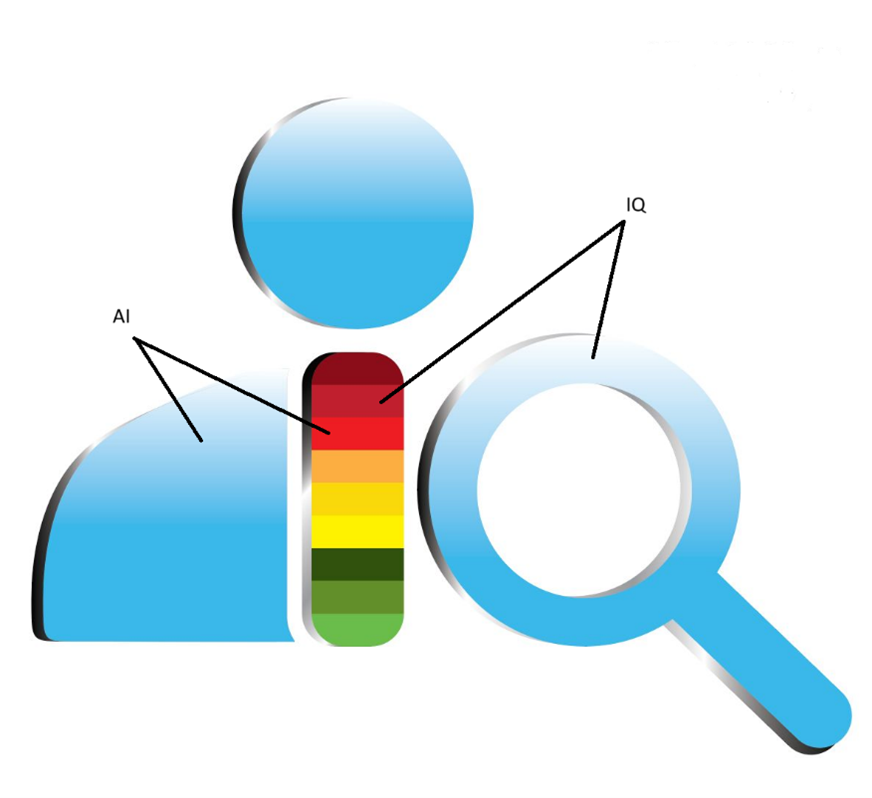
For this article, I will summarize the software for the US FDA market with the focus on bio/pharmaceutical products only. The logo below shows an auditor or inspector with AI and IQ (Intelligence Quotient) built in. The AI’s purpose is to empower IQ of the auditor(s) is the depiction of Figure 17. The AI is at the auditor’s fingertip to ask questions and to get immediate response(s) with references to verify AI’s generated information. This is similar to the FDA’s thinking that “trust but verify” concept when it comes to auditing.
This software is referred to as ieQipTM which stands for intelligent electronic Quality inspection platform.
Are you familiar with the frustration of inefficient or ineffective audit programs but don’t have the resources to hire high qualified consultants? Are you scared by an upcoming FDA inspection which will determine the timeliness of the launch of your new blockbuster? Are you looking for a tool to prevent negative inspection outcome that could put a risk the supply to clients and patients?
Have a look to “ieQipTM”!!! (www.ieQip.com)
Are you equipped for FDA inspections? Yes, “ieQip”!!!
ieQipTM is designed to deliver a regulatory inspection/audit platform which incorporates many concepts from regulatory bodies around the world and structures them in a program that allows professionals to perform in-depth, critical assessments of manufacturing and control processes. In addition, it “measures” the status of compliance, giving you an indication of the criticality of each of the findings identified and the probable classification of the assessment and of the overall maturity of the Quality Management System – to the eyes of the FDA investigator. This is how it works:
- Intelligence is empowered by AI to boost auditor’s intelligence. At the auditor’s fingertips, s/he can pull up the information when there is no readily available answers whether it is regulatory or scientific information.
- The Red, Yellow, and Green is like the streetlights with green means go, yellow means ready to stop, and red means stop
- In FDA terms: red represents OAI (Official Action Indicated), yellow represents VAI (Voluntary Action Indicated), and green represents NAI (No Action Indicated)
- Note: each color has 3 shades indicating the shifting in good or bad direction within that color rating
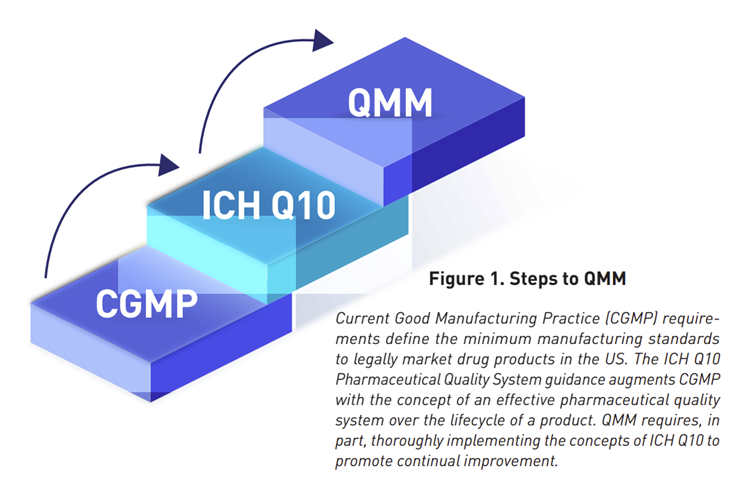
- Remember QMM rating? This software will empower each auditor to rate what they find under a particular citation using scoring of 1-10 (Figure 18) with darker color means the issue being audited is leaning towards worse condition and lighter color means a shift in a better condition as described below
- Red – dark: 1, medium: 2, light: 3
- Yellow – dark: 4, medium: 5, light: 6
- Green – dark: 7, medium: 8, light: 9
3. ieQipTM has the ability for auditor(s) to generate mock FDA-483, discussion items, and report. The AI gives the auditor an intelligence boosts to look for answers quickly with references to verify what was generated by AI.
4. This software can also be used to audit remotely in situations where in person assessment cannot be fulfilled.
5. ieQipTM can be tailored to each manufacturer and/or sponsor’s needs where they have full control of generating assignments to audit or inspect sub-contractors (laboratories, API manufacturers, etc.) as part of their overall quality management system and retain data internally. It can be used to proactively audit or inspect each dosage form or profile (typically inspected by FDA via system-based approach) to continuously assess a company’s quality oversight of their operations in real time.

I developed and will make available training courses covering FDA basic drug inspections, sterile drug inspections, pre-approval inspections, active pharmaceutical inspections, sterile compounding pharmacy inspections, positron emission tomography inspections, and many more. Training can be tailored to your organization’s needs or dosage forms that you manufacture. Trainees will have the opportunity to use the software as a tool to write mock FDA-483, discussion items, and report with supportive evidence to build a red or OAI case.
I trust this approach will help bridging the gap between FDA investigators and industry auditors to allow companies to self-assess their activities and take actions to prevent the risks of import alerts, warning letters, recalls, drug shortages, etc. and ultimately achieve and maintain QUALITY all the time so patients can get the best medicines possible without market supply disruption while incentivizing companies to grow their businesses. Note: this software is not limited to internal use but can also be used by consultants and others who are hired to assess manufacturing operations. This way, the company who has the license to the software can have control over the input data.
Audit of other commodities and other regulatory markets will be further discussed at another time…stay tuned!
________________________________________________________________________________________